Forschungsprojekte
Makroautophagie (nachfolgend Autophagie)
Die Autophagie ist der hauptsächliche lysosomale Prozess für die Beseitigung beschädigter Organellen und den Umsatz von langlebigen Proteinen und Proteinkomplexen. Autophagie findet auf niedrigem Niveau in allen Zelltypen statt; sie wird induziert in Antwort auf Hungerbedingungen und eine Reihe weiterer zellulärer Stresssituationen. In einzelligen Organismen hat sie sich ursprünglich als Überlebensmechanismus entwickelt, damit während Hungerperioden freie Aminosäuren und andere Vorläufermoleküle für essentielle zelluläre Prozesse bereitgestellt werden können. Funktionsstörungen der Autophagie sind mit zahlreichen menschlichen Krankheiten wie Krebs, Neurodegeneration, Muskelschwäche und Fettspeicherstörungen assoziiert und können auch mikrobielle Infektionen fördern. Der Prozess der Autophagie wird in drei Hauptphasen unterteilt, die Autophagosomeninitiation, die Autophagosomenreifung und den lysosomalen Abbau. In der Initiationsphase wird eine Doppellipidmembran de novo generiert; diese wird in eine “tassenförmige” Struktur, das „Phagophor“ in der Hefe Saccharomyces cerevisiae bzw. das „Omegasom“ oder die „Isolationsmembran“ in höheren Eukaryoten erweitert. Diese Struktur expandiert durch den Einbau von Membranlipiden, nimmt für den Abbau bestimmte Fracht auf und wird schließlich zu einem Vesikel, dem Autophagosom geschlossen. Das Autophagosom reift durch die Fusion der äußeren Membran mit dem Lysosom zum Autolysosom. Dann wird die innere Membran des Autophagosoms und sein kompletter Inhalt mittels lysosomaler Hydrolasen abgebaut und die freigesetzten Aminosäuren und andere Degradationsprodukte werden entweder wiederverwertet oder entsorgt (Abb. 2). Viele Proteine mit unterschiedlichen Aktivitäten werden für den regulierten Prozess der Autophagie benötigt. Derzeit sind mehr als 30 “core” Autophagieproteine bekannt.
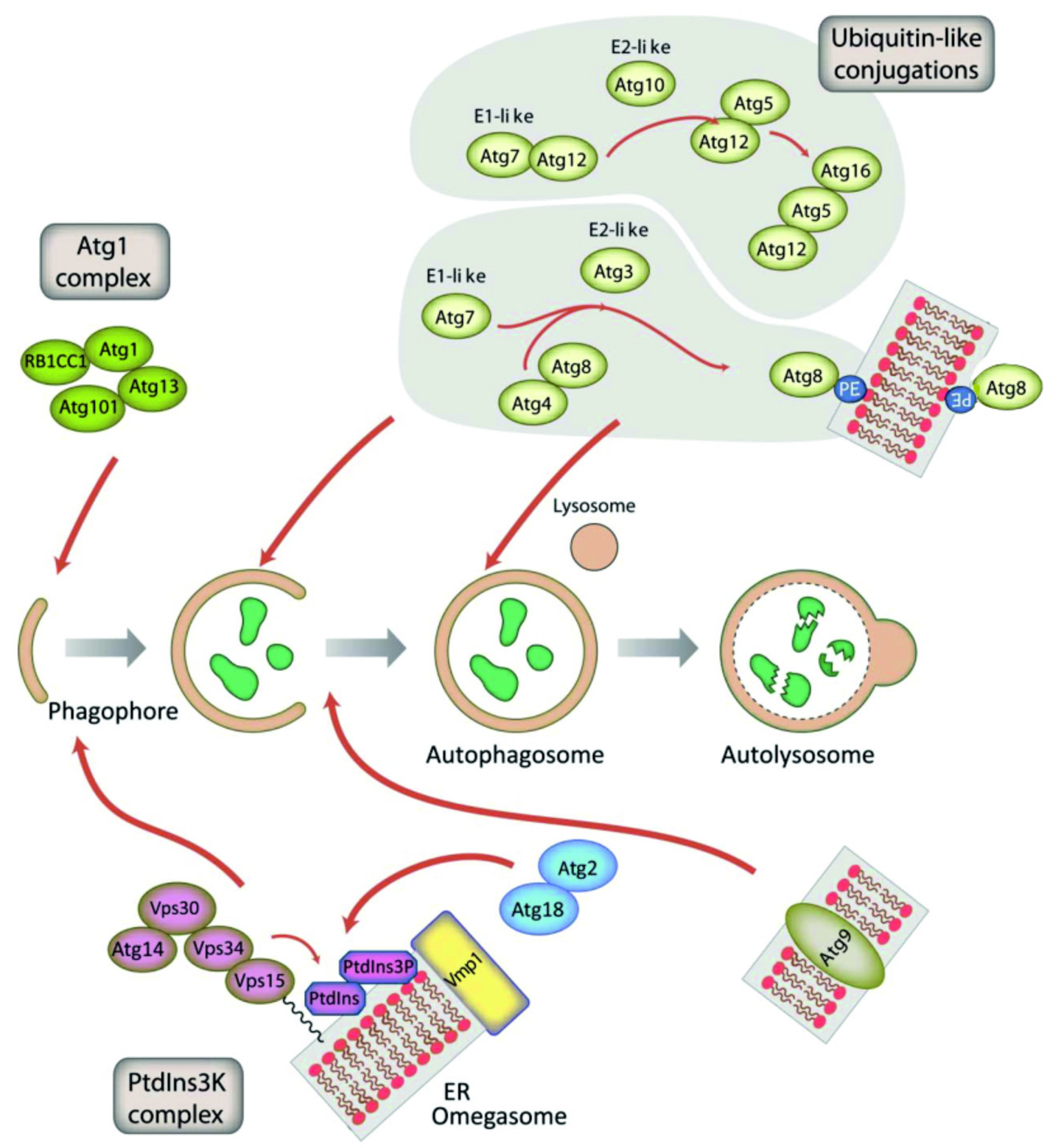
Die Autophagosomenbildung wird durch den Atg1 kinase Komplex und den Klasse III PtdIns3K Komplex induziert. Der letztere Komplex erzeugt das Signallipid PtdIns3P, welches für die Rekrutierung von Atg18 und Atg2 verantwortlich ist. Die Elongation der Membran des Phagophors benötigt zwei Ubiquitin-ähnliche (Ubl) Konjugationssysteme (oben rechts). In der ersten Konjugationsreaktion wird Atg12 kovalent an Atg5 gebunden. Atg12–Atg5 interagiert mit Atg16 und lokalisiert an der Membran des Phagophors. Dort reguliert es die zweite Konjugationsreaktion die Atg8 (LC3 in Säugern) kovalent mit Phosphatidylethanolamin (PE) der expandierenden Phagophorenmembran verknüpft. Atg9-enthaltende Vesikel liefern Membranlipide für die weitere Expansion des Phagophors. Aus Mesquita et al., 2017, Autophagy 13, 24-40, verändert.
Wir untersuchen die Funktionen von ATG5, ATG8a, ATG8b, ATG9, ATG12 and ATG16 und sind insbesondere interessiert an:
- der Initiationsphase der Autophagosomenbildung
- der Verbindung von Autophagie und dem Ubiquitin-Proteasom-System (UPS)
- der LC3 assoziierten Phagozytose (LAP)
- der Rolle der Autophagie bei der Abwehr von Pathogenen
Die Rolle von p97/VCP bei neuro- und myo-degenerativen Erkrankungen
p97, auch bekannt als VCP beim Menschen, TER94 bei Drosophila melanogaster, CdcD bei Dictyostelium discoideum, CDC48 bei Saccharomyces cerevisiae, und VAT bei Thermoplasma acidophilum, ist ein evolutionär hochkonserviertes und ubiquitär exprimiertes Mitglied der Familie der Tripel-A („ATPase Associated with diverse cellular Activities“) ATPasen. Die Domänenstruktur dieser Mg2+-abhängigen ATPase setzt sich aus einer N-terminalen CDC48 Domäne, den D1 und D2 Domänen, die ATP binden und hydrolysieren, und einer C-terminalen Extension zusammen. p97 assembliert in einen ringförmigen hexameren Komplex aus sechs identischen Untereinheiten bei dem die D Domänen den zentralen Zylinder bilden, der von den CDC48 Domänen umgeben ist (Abb. 3). p97 ist an einer Vielzahl zellulärer Prozesse, wie z.B. der Rekonstruktion der Kernmembran, dem Zellzyklus, dem Wiederaufbau des postmitotischen Golgiapparats, der Suppression der Apoptose, der Antwort auf DNA Schäden und der Endozytose, beteiligt. Außerdem wurde gezeigt, das p97 eine zentrale Rolle bei der Proteinqualitätskontrolle spielt.
Bisher wurden mehr als vierzig heterozygote krankheitsverursachende Missensmutationen im humanen p97 beschrieben. Ursprünglich wurde gezeigt, dass p97 Punktmutationen die spät auftretende und langsam fortschreitende Multisystemerkrankung IBMPFD (Inclusion Body Myopathy associated with Paget disease of bone and Fronto-temporal Dementia) verursachen. Mittlerweile wurden vier weitere neurodegenerative Erkrankungen, nämlich ALS (Amyotrophic Lateral Sclerosis), Parkinson, HSP (Hereditary Spastic Paraplegia) und CMT2A2 (Charcot-Marie-Tooth Erkrankung Typ 2), p97 Missensmutationen zugeschrieben. Die exakten molekularen Mechanismen, wie p97 Mutationen diese spät einsetzenden Erkrankungen verursachen, sind weitgehend unverstanden. Jedoch zeigten verschiedene Veröffentlichungen mutationsspezifische Effekte auf p97 Interaktionspartner mit funktionellen Konsequenzen für das endosomale System, den ERAD (endoplasmic reticulum associated degradation of proteins), die Autophagosomenreifung, die ATPase Aktivität oder die Bindung an das 20S Proteasom.
Es wurde auch gezeigt, dass p97 direkt mit Strumpellin (KIAA0196) interagiert. Punktmutiertes Strumpellin verursacht eine schwere, spät-einsetzende und relativ reine Form von HSP (SPG8, OMIM #603563). Strumpellin wurde auch als essentieller Bestandteil des evolutionär hochkonservierten WASH (Wiskott Aldrich Syndrome protein and SCAR homolog) Komplexes identifiziert. Die neurobiologische Relevanz des WASH Proteinkomplexes zeigt sich in der Beobachtung, dass Mutationen in der SWIP (Strumpellin and WASH Interacting Protein) Untereinheit ARID (autosomal recessive intellectual disability) verursachen können.
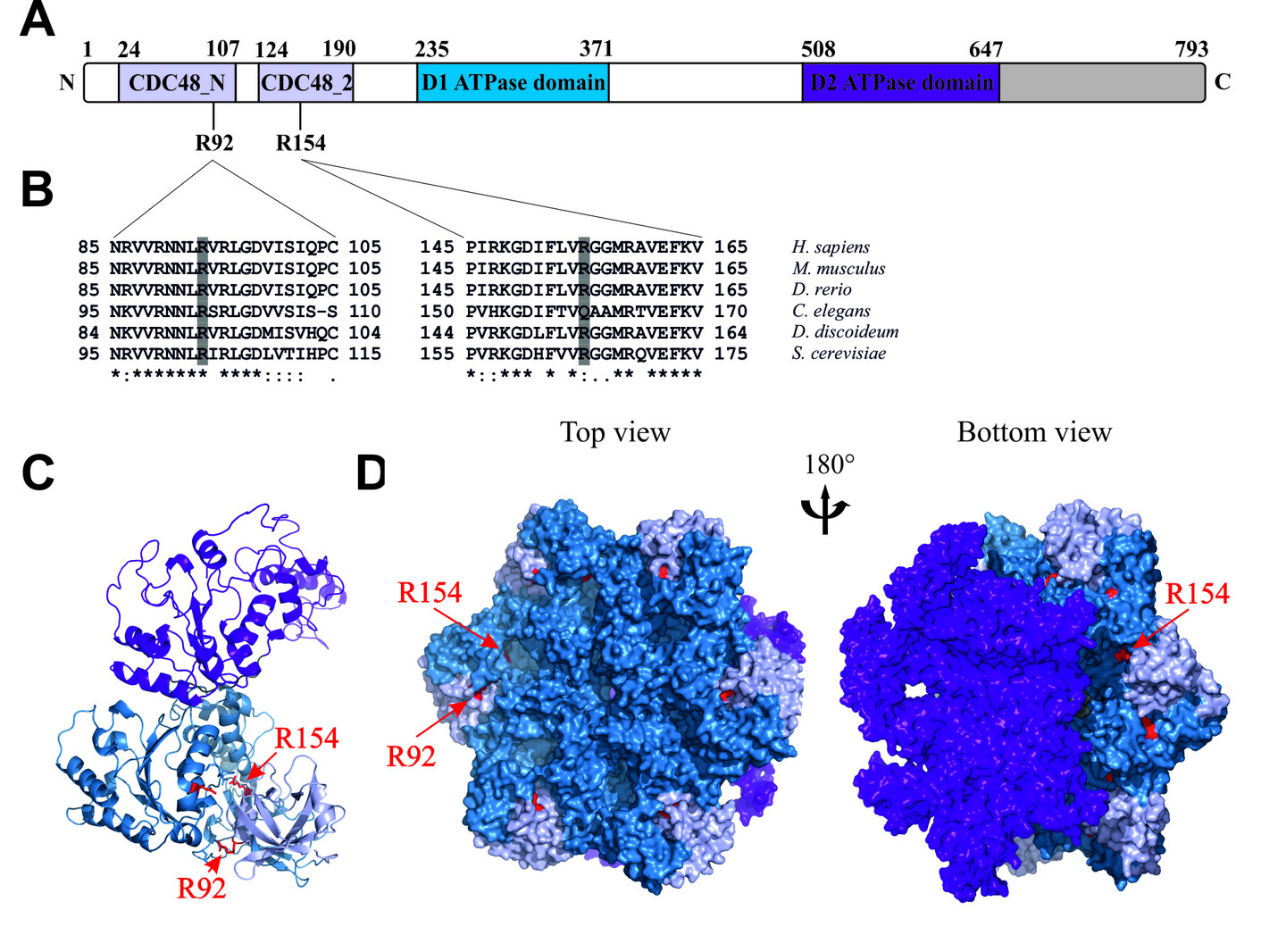
A. Dictyostelium p97 setzt sich zusammen aus einer N-terminalen Region mit den CDC48_N und CDC48_2 Domänen (hellblau), gefolgt von D1 und D2 Domänen (blau und lila, respektive) und einer C-terminalen Extension (grau).
B. Multiples Sequenzvergleich von N-terminalen p97 Sequenzen verschiedener Organismen in der Umgebung der hochkonservierten R92 und R154 Aminosäuren.
C. Sekundärstruktur eines Dictyostelium p97 Monomers.
D. Oberfächendarstellung eines Dictyostelium p97 Hexamers. Die N-Domänen sind hellblau, die D1-Domänen blau, die D2-Domänen lila, und die C-terminalen Extensionen grau dargestellt. Die Aminosäurepositionen von R92 und R154 sind in rot gezeigt.
Von Rijal et al., 2016, Eur J Cell Biol 95, 195-207, verändert.
Wir untersuchen die Funktion von Wildtyp und punktmutiertem p97 in D. discoideum, analysieren die Interaktion mit Strumpellin und anderen bekannten Bindepartner and suchen nach neuen Bindeproteinen. Insbesondere sind wir interessiert an:
- den funktionellen Konsequenzen der p97 R154C, R154H und R92C Mutationen
- der Rolle von Wildtyp und punktmutiertem Strumpellin und seiner Interaktion mit p97
- der Regulation von p97 Funktionen durch Mitglieder der Familie der UBX (ubiquitin regulatory X) Domäne enthaltenden Proteine