Research projects
Macroautophagy (hereafter autophagy)
Autophagy is the major lysosomal pathway for the clearance of damaged organelles and the turnover of long-lived proteins and protein machineries. Autophagy occurs at a basal level in all cell types and is induced in response to starvation and several other cellular stresses. In unicellular organisms it evolved as a survival mechanism to provide free amino acids and other metabolic precursors during starvation. Autophagic dysfunction can lead to numerous human diseases like cancer, neurodegeneration, muscular dystrophy, lipid-storage disorders and may facilitate infections. The autophagic process is subdivided in the autophagosome initiation, maturation and lysosomal degradation phases. In the initiation phase, a membrane sheet is generated de novo, which is transformed into a cup-shaped structure, the phagophore in yeast or the isolation membrane in higher eukaryotes. This structure expands through incorporation of membrane lipids, sequesters cargo and is sealed to form the closed autophagosome. Finally, the autophagosome matures into the autolysosome through fusion of the outer membrane with the lysosome. Then the inner membrane of the autophagosome and its entire contents are degraded by lysosomal hydrolases and the resulting free amino acids and other degradation products are either disposed of or recycled for further use (Figure 2). A plethora of proteins with different activities is required for accurate and regulated autophagy. Currently, more than 30 “core” autophagy proteins are known.
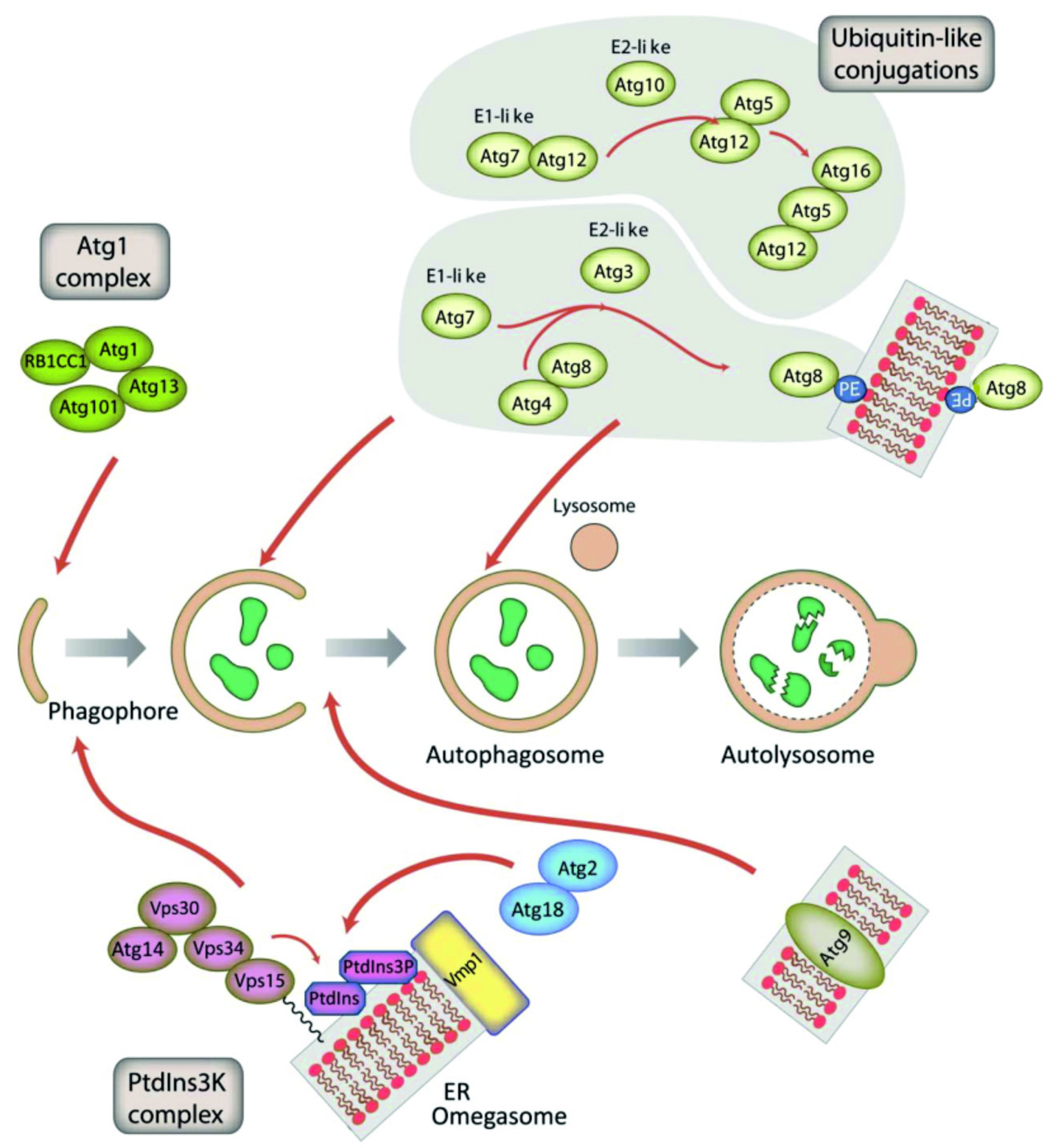
The inductive stage depends on the serine/threonine Atg1 kinase complex and the class III PtdIns3K complex. The latter complex generates the signalling lipid PtdIns3P, which is essential for the recruitment of Atg18 and Atg2. The elongation of the phagophore membrane requires two ubiquitin-like (Ubl) conjugation systems (upper right). In the first conjugation reaction Atg12 is covalently bound to Atg5. Atg12–Atg5 interacts with Atg16 and localizes to the phagophore membrane, from which it regulates the second conjugation reaction that attaches the protein Atg8 (known as LC3 in mammals) to phosphatidylethanolamine (PE) of the expanding phagophore membrane. ATG9-containing vesicles supply membrane for phagophore expansion. From Mesquita et al., 2017, Autophagy 13, 24-40, modified.
We study the functions of ATG5, ATG8a, ATG8b, ATG9, ATG12 and ATG16 in our laboratory and are in particular interested in
- the autophagosome initiation phase
- the link of autophagy to the ubiquitin proteasome system (UPS)
- LC3 associated phagocytosis (LAP)
- the role of autophagy in the defense against pathogens
p97/VCP, a jack of all trades in neuro- and myodegeneration
p97, also known as VCP in Homo sapiens, TER94 in Drosophila melanogaster, CdcD in Dictyostelium discoideum, CDC48 in Saccharomyces cerevisiae, and VAT in Thermoplasma acidophilum, is a ubiquitously expressed, very abundant and evolutionarily highly conserved member of the triple-A (ATPase Associated with diverse cellular Activities) ATPase family. This Mg2+-dependent ATPase has a tripartite structure comprising an N-terminal CDC48 domain followed by the D1 and D2 domains that bind and hydrolyse ATP. p97 assembles into a ring shaped hexameric complex of six identical subunits where the D domains form the central cylinder surrounded by the CDC48 domains (Figure 3). p97 is involved in a plethora of cellular processes such as nuclear envelope reconstruction, cell cycle, postmitotic Golgi reassembly, suppression of apoptosis, DNA damage response, and endocytosis. In addition, it was shown that p97 exerts central roles in several protein quality control pathways.
To date, more than forty heterozygous disease-causing missense mutations have been described in human p97. Initially, it was shown that p97 point mutations cause the late-onset and slowly progressive multi-system disorder IBMPFD (Inclusion Body Myopathy associated with Paget disease of bone and Fronto-temporal Dementia). Meanwhile, four more neurodegenerative disorders, ALS (Amyotropic Lateral Sclerosis), Parkinson’s disease, HSP (Hereditary Spastic Paraplegia) and Charcot-Marie-Tooth disease type 2 (CMT2A2) have been attributed to p97 missense mutations. The exact molecular mechanisms by which p97 mutations cause these late-onset disorders remain elusive. However, an increasing number of reports showed mutation-specific effects on p97 interaction partners with functional consequences on endosomal trafficking, endoplasmic reticulum associated degradation (ERAD) of proteins, autophagosome maturation, ATPase activity or 20S proteasome binding.
VCP was found to directly interact with strumpellin (KIAA0196), which in its mutant form causes a severe and relatively pure motor form of adult-onset HSP (SPG8, OMIM #603563). Furthermore, strumpellin has been identified as a component of the evolutionarily highly conserved WASH (Wiskott Aldrich Syndrome protein and SCAR homolog) complex. The neurobiological relevance of this protein complex is highlighted by the observation that mutations in the WASH complex subunit SWIP (Strumpellin and WASH Interacting Protein) have been attributed to cause familial autosomal recessive intellectual disability (ARID).
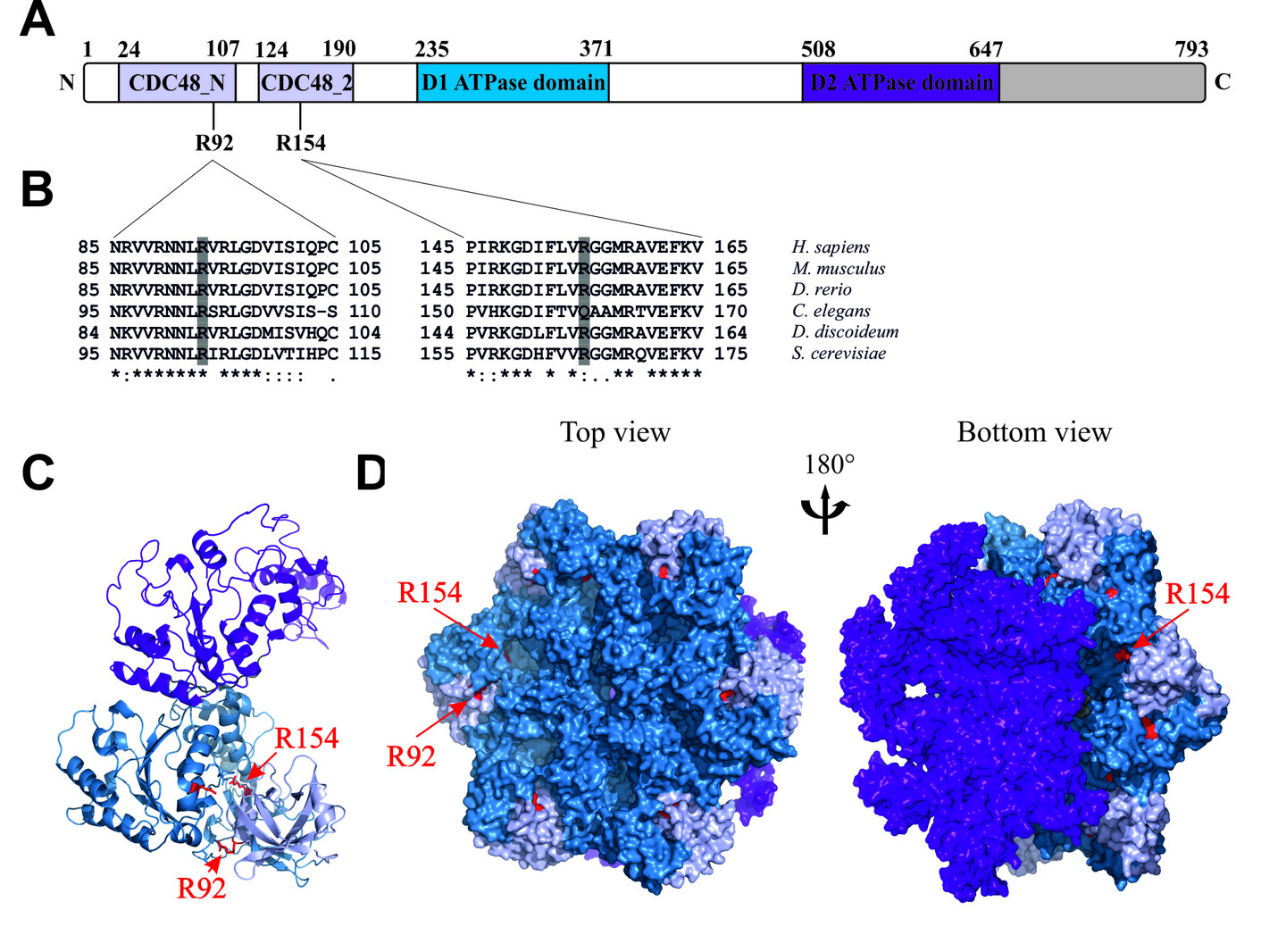
A. Dictyostelium p97 is composed of an N-terminal region comprising the CDC48_N and CDC48_2 domains (light blue), followed by the D1 and D2 domains (blue and purple, respectively) and a C-terminal extension (grey).
B. Multiple sequence alignments of p97 N-terminal sequence stretches flanking the highly conserved R92 and R154 amino acids.
C. Secondary structure of a Dictyostelium p97 monomer.
D. Surface representation of a Dictyostelium p97 hexamer. The N domains are coloured light blue, the D1 domains blue, the D2 domains purple, and the C-terminal extension grey. The positions of R92 and R154 are indicated in red; in the hexamer only positions of a single monomer are lettered.
From Rijal et al., 2016, Eur J Cell Biol 95, 195-207, modified.
We study the function of wild-type and point-mutated p97 in D. discoideum, investigate its interactions with strumpellin and other known binding partners, and search for novel binding proteins. In particular we are interested in
- the functional consequences of the R154C, R154H and R92C mutations
- the role of wild-type and point-mutated strumpellin and its interaction with p97
- the regulation of p97 functions by individual members of the large family of UBX (ubiquitin regulatory X) domain containing proteins