Research projects
Overview
A fundamental principle of cells is their ability to maintain working biological systems in a changing environment. To do so, cells have developed safeguarding mechanisms that monitor the integrity and functionality of their proteome. Cellular surveillance systems sense dysfunction or damage of individual components and elicit adaptive stress responses to ensure cellular or organismal survival by orchestrating their repair, removal and replacement. Failure or deregulation of these protein homeostasis mechanisms is associated with aging as well as severe diseases including cancer, neurodegeneration and inflammatory disorders. We are aiming at a profound mechanistic understanding of protein homeostasis mechanisms controlling the integrity of cellular organelles.
Projects in the lab currently focus on three main aspects:
1.) Intramembrane proteolysis as the new regulatory arm of the ERAD pathway
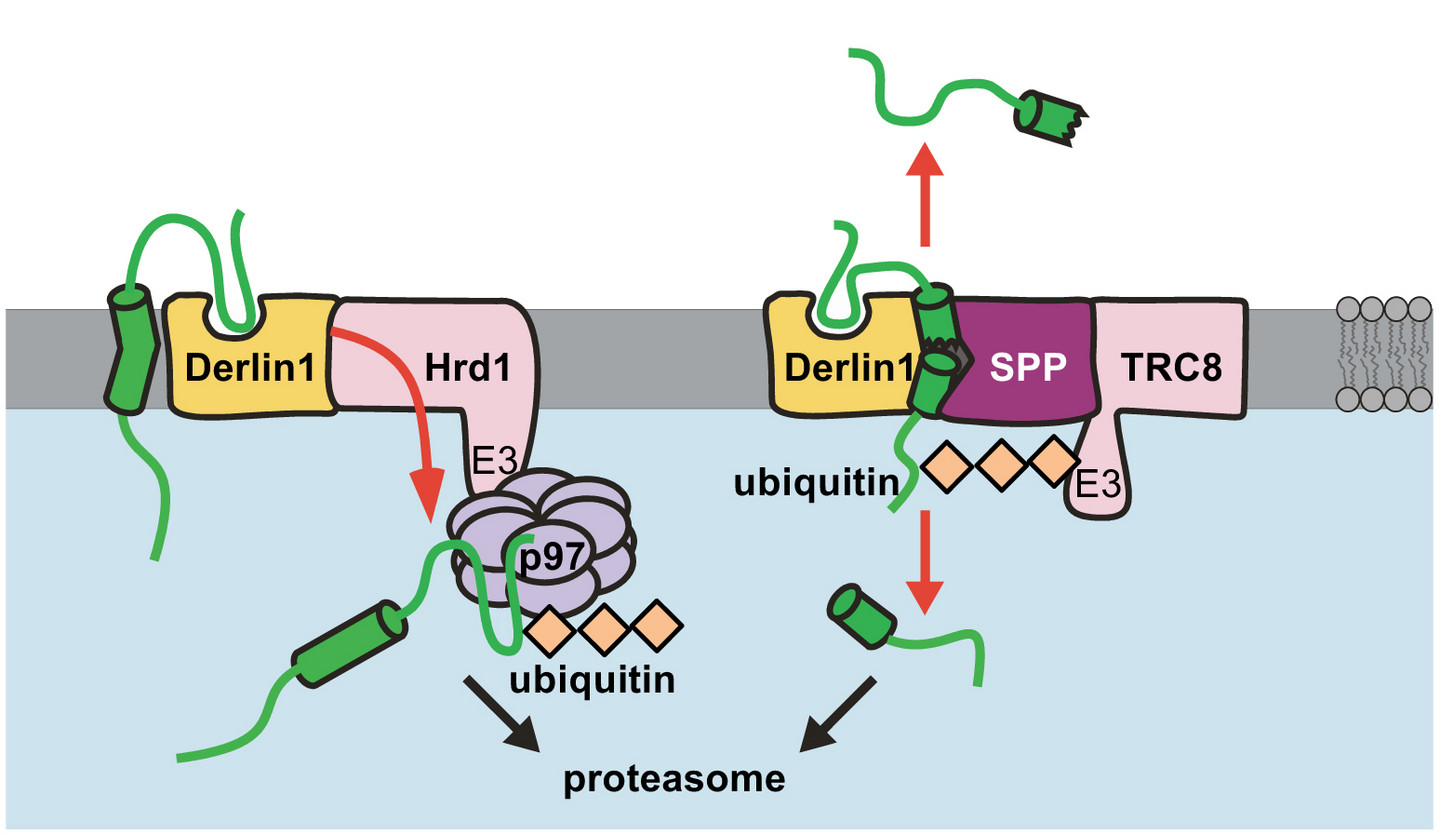
About one third of all mammalian proteins are synthesized in the endoplasmic reticulum (ER), including important cellular proteins such as cell surface receptors and channels. The ER-associated degradation (ERAD) pathway is essentially important to remove misfolded and damaged proteins from the ER to maintain ER protein homeostasis. However, also native proteins can be targeted by ERAD, thereby controlling their abundance. The ERAD machinery forms several parallel pathways that allow recognition and dislocation of a heterogeneous spectrum of substrates. We and others showed that intramembrane proteases and catalytically inactive homologues, the so-called pseudoproteases, serve as central factors in ERAD controlling the abundance and activity of selected membrane proteins. In our lab, we use a combination of cell biology tools, biochemistry and proteomics to study the physiological function and the molecular mechanism of ER-resident proteases and pseudoproteases in protein homeostasis.
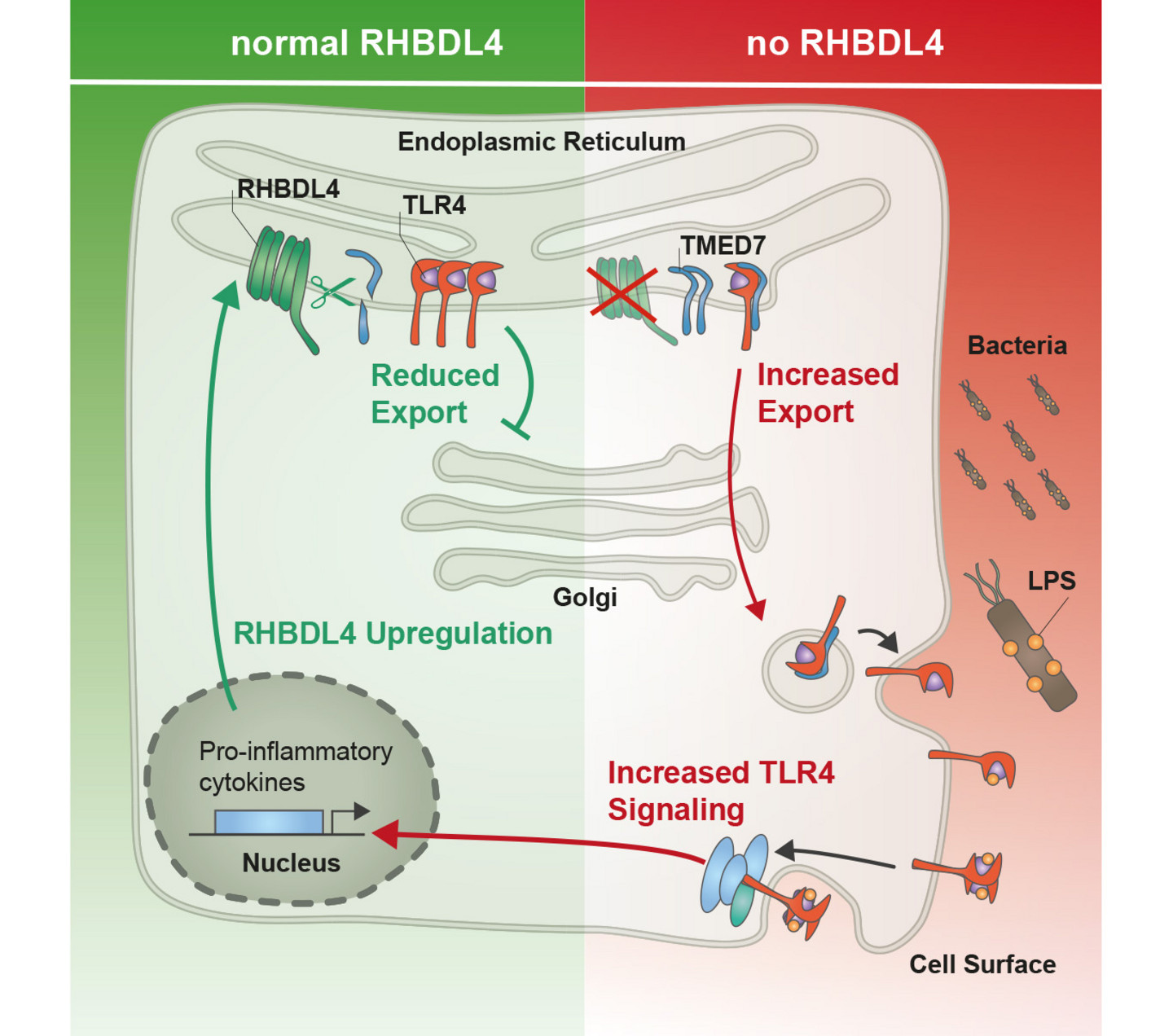
2.) Molecular mechanism of regulated protein secretion
Properly folded proteins are directed from the ER to the Golgi compartment via the so-called COPII-coated vesicles, a process that commonly depends on cargo receptors. We aim to resolve how cargo selection is regulated at the ER exit sites. Recently, we revealed that intramembrane proteolysis tunes the activity of p24 cargo receptors, thereby controlling important plasma membrane proteins such as the immune receptor TLR4.
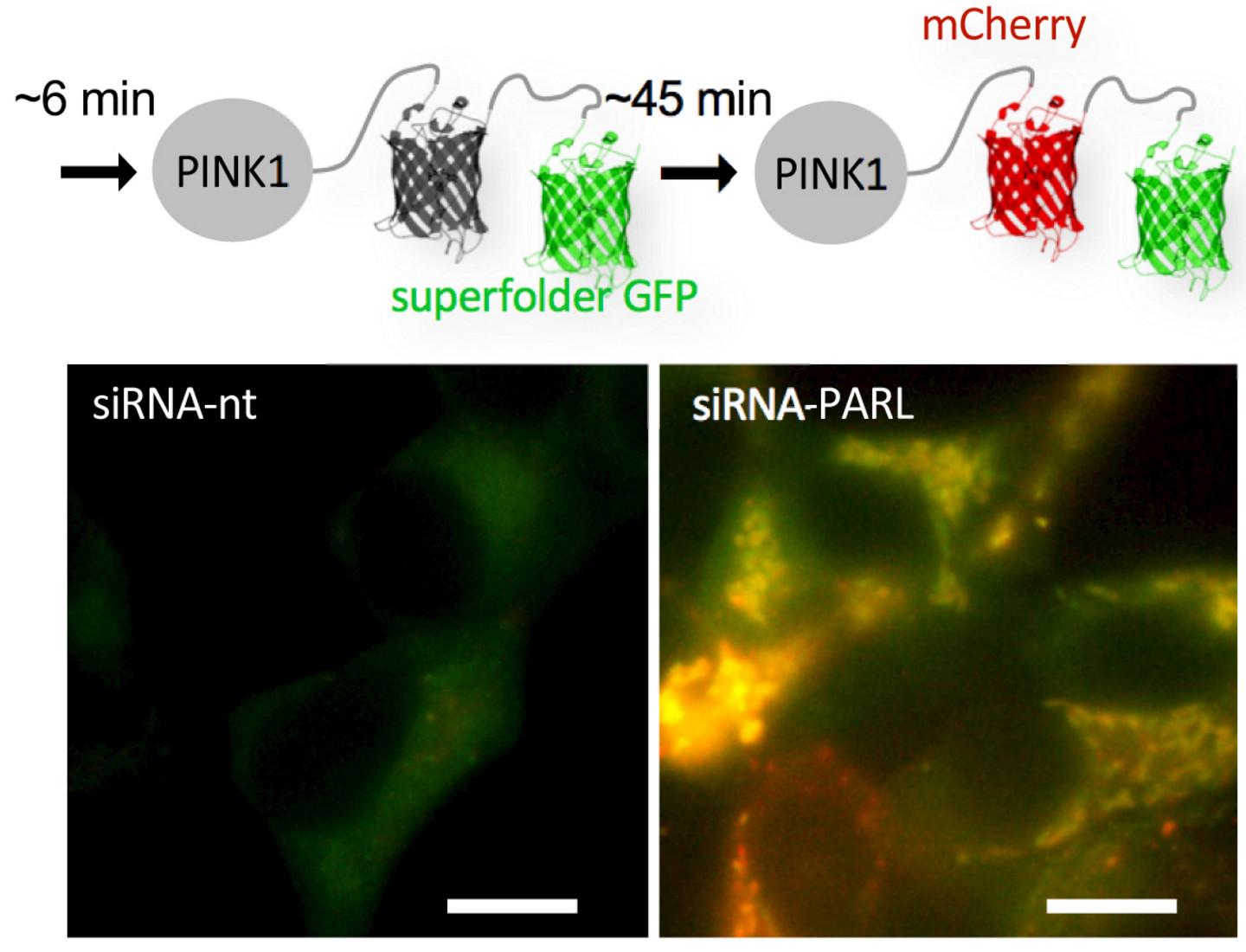
3.) Regulation of mitochondrial protein homeostasis
Mitochondria are highly dynamic organelles required for numerous essential metabolic processes. Mitochondrial dysfunction has severe cellular effects and has been linked to neurodegenerative disorders such as Parkinson’s disease. Our work on mitochondria started when we discovered that the inner membrane protease PARL triggers proteasomal degradation of the serine/threonine kinase PINK1, and thereby controls this central regulator of mitophagy. More recently, we have revealed that the outer membrane dislocase Msp1 in yeast (known as ATAD1/Thorase in humans) synergies with the ERAD E3 ubiquitin ligase Doa10 to control targeting fidelity of tail-anchored proteins. Our main interest is to understand how these mitochondrial protein homeostasis mechanisms interplay with the proteasomal and lysosomal protein turnover.